- Home
- INTERNAL MEDICINE
- Wilson’s Disease


Wilson’s disease is a rare genetic disorder of copper metabolism whereby total body copper is increased with an excess of it being deposited in and causing damage to several organs. It is also known as hepatolenticular degeneration.
Pathophysiology;
Some mutations lead to impaired function of the intracellular copper transporter ATP7B. Usually, copper is absorbed in the stomach and small intestine and taken to the liver, where it is incorporated into ceruloplasmin and secreted into the blood. Excess copper in the body is excreted through the bile. In Wilson’s disease, there is a failure to synthesize caeruloplasmin ( however, in 5%of patients, caeruloplasmin levels are within the normal range).
Impaired biliary excretion of copper leads to copper accumulation in several organs, notably the liver, basal ganglia of the brain, cornea, kidneys, and the skeleton. The liver is affected first.
Clinical presentation
- Symptoms can occur between the ages 5 and 45.
- Children present initially with the hepatic disease between 9 and 13 years
- Adolescents and adults present more often with neurologic symptoms
1. Liver disease.
They may present with;
- Acute Hepatitis, sometimes recurrent
- Fulminant liver failure leads to copper release into the bloodstream, causing massive hemolysis and renal tubulopathy.
- Chronic Hepatitis, presenting with cirrhosis; liver failure, and portal hypertension
- Jaundice
- Hepatomegaly
- Splenomegaly
- Mental status change due to hepatic encephalopathy
2. Neurologic disease
May present with;
- Tremor, Choreoathetosis, Parkinsonism, Dystonia, Dementia, dysarthria, ataxia, drooling.
- Others include seizures, hyperreflexia, myoclonia, urinary incontinence, and autonomic dysfunction.
- Unusual clumsiness for age is an early feature.
- It can be prevented by treatment started following diagnosis of the liver disease phase.
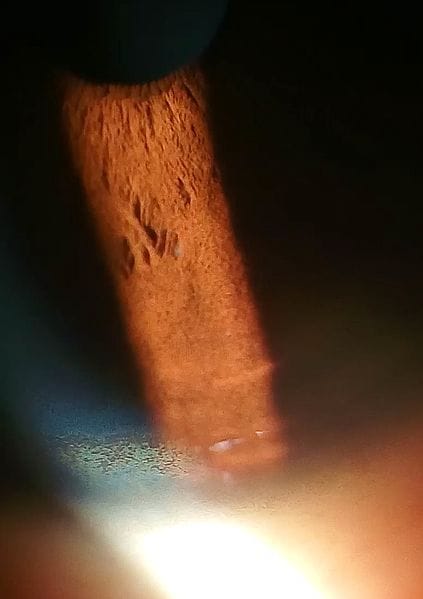
3. Kayser-Fleischer rings
- They are a finding in 60% of adults with the disease. It’s the most critical clinical clue to the disease.
- They are due to fine, pigmented, granular deposits of copper on the Descemet’s membrane of the cornea. They are characterized by greenish-brown discolouration of the corneal margin; they first appear at the upper periphery.
- Often detected by slit-lamp examination, but sometimes are visible without a slit lamp examination.
- Usually, disappear with treatment
Investigations
- Serum caeruloplasmin levels- low. Advanced liver failure may, however, lead to low caeruloplasmin levels. Sometimes the level is normal
- Free copper concentration- may be elevated in the serum, urine, and the liver
- 24-hour urinary copper while administering D penicillamine; a level of more than 25 micro mols in 24 hours is diagnostic
- Liver function tests-elevated aminotransferases
- CBC- thrombocytopenia with splenomegaly
- Coomb’s test- coomb’s negative hemolytic anaemia
- Abdominal imaging- hepatosplenomegaly
- MRI or CT scan for neurologic disease
Management
General-
- low copper diets,
- regular checkups with liver biochemical tests every six months for stable disease
1. Penicillamine is the drug of choice. It’s a copper-binding agent.
- Dose- 1 to 4 mcg per day
- Treatment must continue for life
- dose reduction in remission, but take care to avoid copper re accumulation
- Toxic effects- rash, lupus-like syndrome, protein-losing enteropathy, and bone marrow depression. Give trientine dihydrochloride and zinc as alternatives
2. Liver transplant-
- advanced cirrhosis, fulminant liver failure
3. Investigate and treat siblings and children of patients with Wilson’s disease, even if asymptomatic.
Prognosis
Excellent in all but those with advanced disease and those who presentation with fulminant liver failure and hemolysis